



Содержание
Перейти к:
https://doi.org/10.35266/2949-3447-2025-1-9
Перейти к:
Однородительская дисомия – аномалия, при которой обе гомологичные хромосомы наследуются от одного родителя. Патологические эффекты рассматриваемой аномалии связаны с нарушениями импринтинга и потерей гетерозиготности. Классические методы диагностики, такие как микросателлитный анализ и хромосомный матричный анализ, имеют ограничения при диагностике аномалии в неполных семьях и мозаичных случаях. Цель данного исследования – оценка потенциальной эффективности метода диагностики однородительских дисомий хромосом и нарушений импринтинга на основании профилирования метилирования ДНК по данным секвенирования нового поколения. В частности, был модифицирован метод пробоподготовки библиотек геномной ДНК с использованием метилзависимой эндонуклеазы GlaI в сочетании с эндонуклеазой рестрикции Bst2UI для увеличения информативности анализа, а также разработан биоин-форматический алгоритм обработки данных. Метод был протестирован на клиническом случае реципрокной транслокации (3;19)(q12;q13.3) по материнской линии с подозрением на сегментарную однородительскую дисомию у пробанда. Модификация протокола пробоподготовки позволила достичь охвата около 5 млн сайтов метилирования. Биоинформатический анализ включал определение статуса метилирования клинически значимых областей контроля импринтинга и поиск областей гомозиготности. Метод позволил охватить 2/3 потенциальных областей контроля импринтинга. Признаков однородительской дисомии у пробанда обнаружено не было, что согласуется с результатами хромосомного матричного анализа. Несмотря на то что подход представляет собой экономически эффективную альтернативу полногеномному бисульфитному секвенированию, остаются нерешенными проблемы с нормализацией получаемых относительных уровней метилирования. В дальнейшем планируется провести валидацию разработанного на биологических образцах с подтвержденными случаями однородительской дисомии, чтобы сделать однозначный вывод о его пригодности для идентификации рассматриваемых аномалий.
Сучко П.А., Некрасова Д.А., Донников М.Ю., Глотов О.С., Данилов Л.Г. МЕТОД СЕКВЕНИРОВАНИЯ И АЛГОРИТМ АНАЛИЗА ПРОФИЛЕЙ МЕТИЛИРОВАНИЯ НА ОСНОВАНИИ ДАННЫХ NGS-СЕКВЕНИРОВАНИЯ ДЛЯ ВЫЯВЛЕНИЯ ОДНОРОДИТЕЛЬСКИХ ДИСОМИЙ. Вестник СурГУ. Медицина. 2025;18(1):73-80. https://doi.org/10.35266/2949-3447-2025-1-9
Suchko P.A., Nekrasova D.A., Donnikov M.Yu., Glotov O.S., Danilov L.G. SEQUENCING METHOD AND ALGORITHM FOR ANALYZING METHYLATION PROFILES BASED ON NGS DATA TO IDENTIFY UNIPARENTAL DISOMY. Vestnik SurGU. Meditsina. 2025;18(1):73-80. (In Russ.) https://doi.org/10.35266/2949-3447-2025-1-9
Однородительская дисомия (ОРД) хромосомы – аномалия, при которой обе гомологичные хромосомы или хромосомные сегменты унаследованы от одного родителя [1]. Хотя подобные случаи зафиксированы для всех аутосом человека и Х-хромосомы [2], в качестве клинически значимых рассматриваются ОРД хромосом 6, 7, 11, 14, 15, и 20 [3]. По аллельному соотношению выделяют два типа данных аномалий: при изодисомии наследуется две идентичные копии хромосомы одного родителя, а при гетеродисомии наследуются две гомологичные хромосомы одного родителя [4].
Патологические последствия ОРД обусловлены потерей гетерозиготности, а также нарушениями геномного импринтинга, что является причиной синдромов Ангельмана, Прадера – Вилли, Беквита – Видемана, Сильвера – Рассела и др. [5]. Частота ОРД оценивается примерно в 1 на 2000 родов [6]. Помимо врожденной также встречается приобретенная ОРД в соматических клетках, что является распространенной особенностью различных видов рака [7].
Характерной особенностью синдромов, связанных с ОРД, является множественность молекулярных причин заболевания: помимо рассматриваемой аномалии значительный вклад вносят делеции, мутации и аберрантное метилирование в областях контроля импринтинга (ICR), поэтому точное установление этиологии играет в этих клинических случаях ключевую роль [8]. Кроме того, тестирование на ОРД назначается при обнаружении структурных перестроек хромосом 14 и 15, а также при идентификации моносомного мозаицизма или трисомии в ходе пренатальной диагностики [1][9].
Классическим методом диагностики ОРД является микросателлитное тестирование, но необходимость биоматериала обоих родителей пробанда (трио-анализ), трудности при выявлении сегментарных дисомий и анализе мозаичных случаев ограничивают его эффективность [1][5]. В последние годы преобладающими методами становятся хромосомный матричный анализ (ХМА) и экзомное секвенирование благодаря своей производительности и возможности выявления областей гомозиготности (ROH) различной протяженности, однако мозаицизм и потребность в родительском материале по-прежнему остаются значительной проблемой [10][11]. В качестве дополнительных инструментов используются метил-специфическая ПЦР и метил-специфическая мультиплексная амплификация лигированных зондов для оценки статуса метилирования ICR [12]. Наибольшая эффективность достигается при комбинировании различных подходов, что существенно повышает стоимость тестирования и увеличивает сроки диагностики.
Перспективным инструментом для выявления однородительских дисомий и постановки дифференцированного диагноза при болезнях геномного импринтинга может стать сочетание профилирования метилирования ДНК и оценка ROH в рамках одного тестирования для выявления всего возможного комплекса аномалий, приводящих к дисрегуляции ICR. На текущий момент подобный подход был реализован с использованием адаптивного нанопорового секвенирования на платформе Oxford Nanopore и позволил идентифицировать различные молекулярные причины синдромов Ангельмана и Прадера – Вилли, в т. ч. однородительскую дисомию, однако его недостатком является чрезвычайная дороговизна [13][14]. В качестве альтернативы могут рассматриваться методы, основанные на аффинном обогащении метилированных последовательностей или расщеплении геномной ДНК метил-чувствительными либо метилзависимыми эндонуклеазами, но на текущий момент в литературе отсутствуют свидетельства их применения для диагностики ОРД.
В рамках данной работы для разработки подхода к комплексному анализу ROH и паттернов метилирования ICR была осуществлена модификация метода высокопроизводительного секвенирования, основанного на фрагментации геномной ДНК метилзависимой эндонуклеазой GlaI (R (5mC)↑GY), ограничением которого являлось малое количество охватываемых RCGY-сайтов [15].
Цель – оценка потенциальной эффективности метода диагностики однородительских дисомий хромосом и нарушений импринтинга на основании NGS-профилирования метилирования ДНК с использованием метилзависимой эндонуклеазы GlaI.
Выбор рестриктаз для последовательного гидролиза. Для расширения пула секвенируемых фрагментов был выбран ряд рестриктаз, нечувствительных к метилированию и с частым сайтом узнавания, не перекрывающим сайт RCGY: Bst2UI (CC↑WGG), HaeIII (GG↑CC), RsaI (GT↑AC). Для выбранных ферментов, а также для GlaI было выполнено расщепление референсного генома hg38 in silico с использованием программного пакета ddRADseqTools [16]. Полученные фрагменты в формате fasta картировались на референсный геном с помощью BWA-MEM. Далее картированные фрагменты с помощью HTseq [17] сопоставлялись с аннотациями CCDS, промоторов, повторов и энхансеров, загруженных через USCS Table Browser.
Подбор пациентов и условий фрагментации геномной ДНК. Для отладки методики пробоподготовки были взяты образцы периферической крови от 7 пациентов-родственников, среди которых присутствовало 2 пары мать – ребенок. Отличительной особенностью рассматриваемой когорты является наличие семейной реципрокной транслокации (3;19) (q12; q13.3) по материнской линии, при этом у пробанда, мальчика 7 лет, страдающего задержками психомоторного и психоречевого развития, гиперактивностью, дефицитом витамина D, неуточненной энцефалопатией, была заподозрена врачом-генетиком сегментарная однородительская дисомия одной из затронутых транслокацией хромосом. Наличие транслокации подтверждено кариотипированием.
Геномная ДНК выделялась из периферической крови с использованием набора Nucleic Acid Extraction Kit (BGI, Китай). 1000 нг геномной ДНК фрагментировали 10 е. а. метилзависимой эндонуклеазой GlaI (SibEnzyme, Россия) в течение 2 часов, далее производили очистку гидролизата с использованием набора Cleanup Mini (Евроген, Россия). Далее параллельно тестировался дополнительный гидролиз с 10 е. а. Bst2UI, HaeIII и RsaI (SibEnzyme, Россия) в течение 4 часов. Фрагментный состав полученных смесей анализировался на станции автоматизированного электрофореза TapeStation 4150 (Agilent Technologies, США). Отбор фрагментов в целевом диапазоне длин 100–600 п. н. осуществлялся для смеси GlaI + Bst2UI с помощью магнитных частиц Hieff NGS DNA Selection Beads (Yeasen, Китай).
Подготовка библиотек и секвенирование. При подготовке библиотек использовали 100 нг продукта последовательного гидролиза ДНК GlaI и Bst2UI. Пробоподготовка осуществлялась согласно протоколу производителя набора MGIEasy Universal DNA Library Prep Set (MGI, Китай). Секвенирование производилось на платформе DNBSEQ-G400 (MGI, Китай) c длиной прочтений 110 п. н.
Обработка данных секвенирования. Предварительная фильтрация прочтений с Q ≥ 30 и обрезка адаптеров DNBSEQ выполнялась с использованием Trimmomatic. Контроль качества прочтений осуществлялся с помощью утилиты FastQC. Далее производилось картирование прочтений c использованием BWA-MEM. Из файлов bam с помощью специально разработанного скрипта на Python удалялись прочтения, не соответствующие сайту узнавания GlaI.
Подсчет количества прочтений, пересекающих интервалы геномных аннотаций, осуществлялся с помощью HTseq. Координаты кандидатных ICR были загружены через геномный браузер HumanICR [18]. Для образцов, составляющих пару мать – ребенок, было выполнено аннотирование SNP с помощью snpEff, полученные vcf-файлы тестировались на однородительскую дисомию с использованием altAFplotter [19]. Отсутствие протяженных участков потери гетерозиготности (> 3 000 000 п. н.) подтверждено результатами ХМА экзонного уровня на генетическом анализаторе ГЕНОСКАН 3000 (ООО «Геноскан», Россия).
Получено согласие этического комитета Сургутского государственного университета на публикацию материала на заседании от 26 апреля 2024 г.
При расщеплении эталонного генома in silico эндонуклеазами GlaI, HaeIII, Bst2UI и RsaI было подсчитано число фрагментов, потенциально образующихся в ходе гидролиза. Полученные данные представлены в табл. 1. Можно заметить, что наибольшее количество потенциальных фрагментов образуется с Bst2UI, и эта рестриктаза с наибольшей вероятностью способствует расширению пула захватываемых фрагментов при последовательном гидролизе после GlaI.
Таблица 1
Число рестрикционных фрагментов in silico
Эндонуклеаза | GlaI | Bst2UI | HaeIII | RsaI |
Сайт узнавания | R (5mC) ↑GY | CC↑WGG | GG ↑CC | GT↑AC |
Общее число фрагментов | 8 009 924 | 10 721 268 | 9 306 384 | 5 440 264 |
Число фрагментов 100–600 п. н. | 3 590 852 | 5 050 756 | 4 145 252 | 2 546 112 |
Примечание: составлено авторами.
Ограничением расщепления in silico является отсутствие учета статуса метилирования расщепляемых сайтов, что в случае GlaI приводит к несоответствию реальному паттерну расщепления. На рис. 1 показано распределение фрагментов in silico для GlaI и Bst2UI.
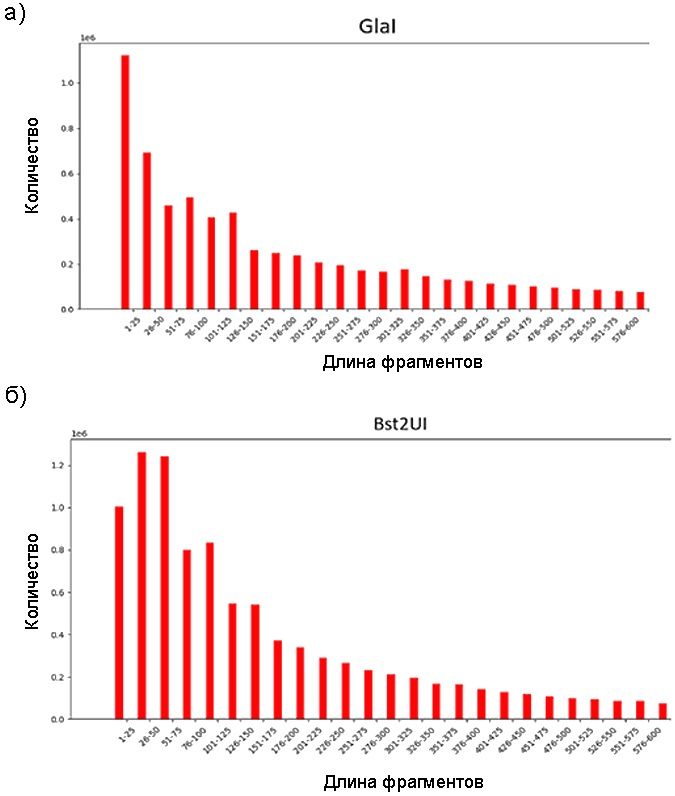
Рис. 1. Распределение фрагментов in silico:
а) распределение смоделированных фрагментов GlaI;
б) распределение смоделированных фрагментов Bst2UI
Примечание: изображение авторов.
На рис. 2 показано реальное распределение фрагментов при последовательном двойном гидролизе и при обычном расщеплении геномной ДНК эндонуклеазой GlaI (рис. 2 г). Наблюдаемое для GlaI распределение не меняется при увеличении времени инкубации до 16 часов и согласуется с более ранней работой M. A. Abdurashitov и соавт. [15]. Таким образом, двойной последовательный гидролиз геномной ДНК с GlaI и Bst2UI позволяет значительно расширить пул фрагментов для секвенирования. В то же время такой подход связан с сильным смещением в сторону CpG-бедных участков генома и вероятной потерей информации о регуляторных последовательностях, богатых CpG (в частности, промоторах и энхансерах).
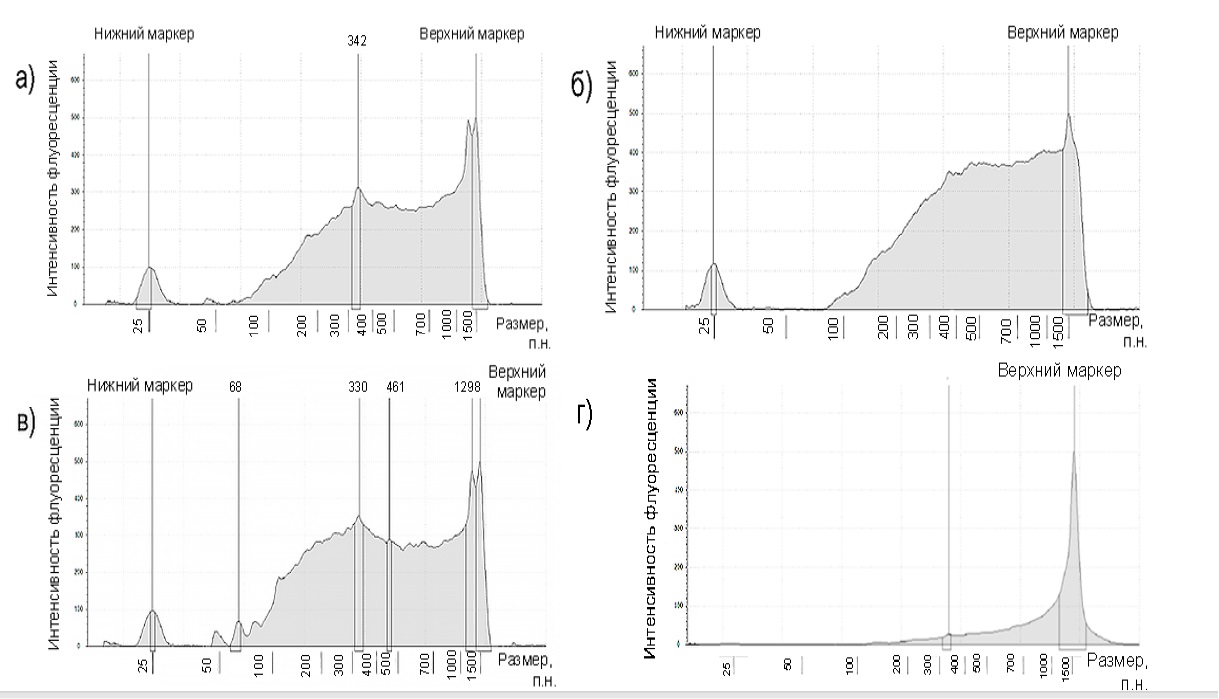
Рис. 2. Реальное распределение фрагментов (электрофореграмма с прибора TapeStation 4150):
а) распределение фрагментов после обработки геномной ДНК эндонуклеазами GlaI и HaeIII;
б) распределение фрагментов после обработки геномной ДНК эндонуклеазами GlaI и RsaI;
в) распределение фрагментов после обработки геномной ДНК эндонуклеазами GlaI и Bst2UI;
г) распределение фрагментов после обработки геномной ДНК эндонуклеазой GlaI
Примечание: изображение авторов.
В ходе секвенирования было получено в среднем 75,7 млн прочтений на образец (Q ≥ 30), из которых 95 % корректно картировались на референсный геном. В то же время лишь 25 % из этих прочтений прошли фильтр по сайту узнавания GlaI, что указывает на присутствие в смеси значительного количество фрагментов, являющихся продуктами расщепления только рестриктазы Bst2UI. Таким образом, по отношению к задаче определения сайтов метилирования 3/4 данных секвенирования являются нецелевыми. В то же время применительно к диагностике ОРД они могут способствовать увеличению чувствительности за счет дополнительных SNP, учитываемых altAFplotter при поиске ROH.
В результате подсчета индивидуальных сайтов метилирования было выявлено около 5 млн охватываемых CpG в составе RCGY-сайтов со средним покрытием 4-х из примерно 7,3 млн сайтов RCGY в референсном геноме. Ранее в работе M. A. Abdurashitov и соавт. удавалось охватить до 3,5 млн сайтов RCGY [15]. Таким образом, применение дополнительной рестриктазы позволяет значительно увеличить информативность анализа как для анализа паттернов метилирования, так и для детекции SNP. Однако подобная схема пробоподготовки приводит к увеличению затрат на секвенирование, особенно при необходимости достижения глубины 30-x за счет расширения покрываемой части референсной генома с 10–12 до 40 %.
Для отфильтрованных по сайту узнавания GlaI прочтений и фрагментов in silico было выполнено сопоставление с геномными аннотациями, результаты представлены в табл. 2.
Таблица 2
Распределение прочтений в эксперименте/фрагментов in silico по геномным элементам (в %)
Промоторы | CCDS | Энхансеры | Гены | Повторы | ||||
от общего числа прочтений | от числа элементов аннотации | от общего числа прочтений | от числа элементов аннотации | от общего числа прочтений | от числа элементов аннотации | от общего числа прочтений | от общего числа прочтений | |
Реальные прочтения | 1,1 | 12,4 | 13,3 | 85,5 | 0,9 | 18,0 | 15,7 | 39,1 |
Фрагменты GlaI in silico | 0,38 | 35,5 | 2,4 | 46,3 | 0,017 | 35,6 | 2,8 | 37,3 |
Фрагменты GlaI + Bst2UI in silico | 0,18 | 18,5 | 1,70 | 40,9 | 0,015 | 26,4 | 4,47 | 48 |
Примечание: составлено авторами.
По данным табл. 2 видно, что значительную часть прочтений составляют повторяющиеся элементы генома. По сравнению с фрагментами GlaI in silico, при секвенировании продуктов двойного последовательного гидролиза происходит потеря существенной доли информации о регуляторных элементах генома (в частности, промоторов и энхансеров). Вероятно, это связано с высокой плотностью расположения сайтов гидролиза GlaI и Bst2UI в этих участках.
На рис. 3 приведен пример покрытия хромосомы 15 как наиболее часто подверженной возникновению ОРД, а также импринтированного локуса SNRPN-SNURF.
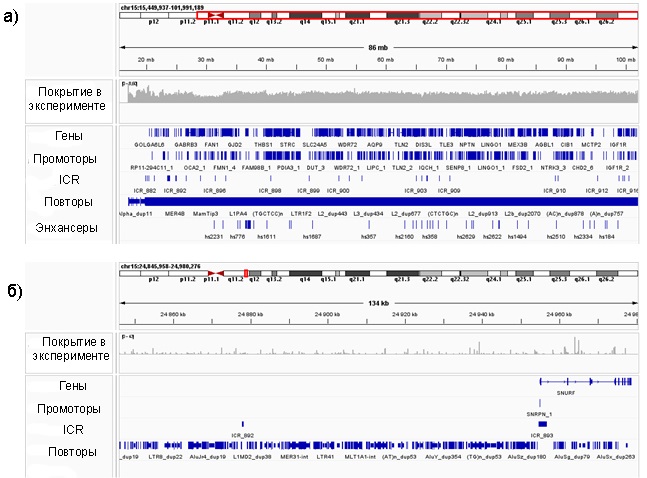
Рис. 3. Визуализация прочтений хромосомы 15 у пробанда в эксперименте:
а) карта покрытия длинного плеча хромосомы 15;
б) карта покрытия импринтированного локуса SNRPN-SNURF
Примечание: составлено авторами.
Для данных, полученных в ходе апробации метода секвенирования, характерна неравномерность покрытия (рис. 3 а). Учитывая недостаточность количества прочтений на покрытую часть генома, на основе пилотного набора данных нет возможности сделать достоверный вывод о статусе ICR (рис. 3 б), вносящих вклад в фенотипическое проявление ОРД. С учетом объема данных секвенирования в пилотном запуске, для обеспечения высокого покрытия (около 30-х) необходимо около 300 млн прочтений, что делает метод в разы более экономичным, чем полногеномное бисульфитное секвенирование, являющееся золотым стандартом для анализа профилей метилирования. Следует отметить, что полученные прочтения охватывают сайты метилирования в 993 кандидатных ICR из 1488. При этом среди клинически значимых охвачены ICR, участвующие в патогенезе синдромов Прадера – Вилли, Ангельмана, Сильвера – Рассела и транзиторного неонатального сахарного диабета. По-видимому, сдвиг в сторону CpG-бедных участков генома оказывается не столь критичным для анализа ICR, что согласуется с данными о перекрытии 850 000 CpG в микрочипе Infinium Methylation EPIC (llumina, США) сайтов CpG в ICR всего на 7 % [20]. Поскольку метилзависимые эндонуклеазы позволяют получать информацию об относительном уровне метилирования, а не абсолютном, как при бисульфитном секвенировании, для достоверного определения статуса метилирования ICR критическое значение имеет нормализация полученных данных, либо сопоставление уровней метилирования в ICR в контрольной группе здоровых людей с уровнями метилирования у пациентов с ОРД.
Что касается анализа уровней метилирования в ICR на хромосомах 3 и 19, затронутых в данном клиническом случае реципрокной транслокацией, то при сопоставлении данных секвенирования пробанда и родственников без хромосомной перестройки установлена статистически значимая (p > 0,05) корреляция, (p = 0,79) по Спирмену, что указывает на высокое сходство паттернов метилирования у больного и здоровых родственников и вероятное отсутствие сегментарной ОРД. Кроме того, при анализе vcf-файлов не было выявлено протяженных областей гомозиготности, что также может указывать на отсутствие ОРД у пробанда.
В ходе работы была проведена апробация подхода к диагностике однородительских дисомий на основе анализа профилей метилирования и областей гомозиготности по данным NGS в рамках одного тестирования. В рассмотренном клиническом случае не было получено данных, подтверждающих наличие ОРД у пробанда, что также соотносится с результатами хромосомного матричного анализа. При этом разработанный подход потенциально обладает большей чувствительностью, чем ХМА, за счет возможности оценки уровней метилирования в ICR, нарушающихся при ОРД и определяющих их фенотипическое проявление, а также анализа большего числа полиморфных генетических вариантов. В качестве недостатков подхода можно отметить сложность интерпретации данных секвенирования, высокие затраты биоматериала, а также высокую стоимость анализа, которая, тем не менее, почти вдвое ниже стоимости полногеномного секвенирования.
В дальнейшем планируется проведение дополнительного испытания разработанного подхода на образцах от пациентов с подтвержденными однородительскими дисомиями и болезнями геномного импринтинга, однако основной трудностью в проведении подобного исследования является низкая частота данных заболеваний (менее 1 на 10 000 чел.), сложность молекулярно-генетического тестирования и низкая распространенность соответствующих методов диагностики. В случае успешности такого испытания описанный подход может стать инструментом первой линии для анализа клинических случаев с подозрением на ОРД и нарушения импринтинга, на основании результатов которого могут быть назначены более простые методы точечной диагностики наиболее вероятных молекулярных аномалий.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Conflict of interest. The authors declare no conflict of interest.
1. Del Gaudio D., Shinawi M., Astbury C. et al. Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG) // Genetics in Medicine. 2020, Vol. 22, no. 7. P. 1133–1141. https://doi.org/10.103 8/s41436-020-0782-9.
2. Liehr T. 2025. Cases with uniparental disomy. URL: https://cs-tl.de/DB/CA/UPD/0-Start.html (дата обращения: 13.01.2025).
3. Benn P. Uniparental disomy: Origin, frequency, and clinical significance // Prenatal Diagnosis. 2021. Vol. 41, no. 5. P. 564–572. https://doi.org/10.1002/pd.5837.
4. Eggermann T., Soellner L., Buiting K. et al. Mosaicism and uniparental disomy in prenatal diagnosis // Trends in Molecular Medicine. 2015. Vol. 21, no. 2. P. 77–87. https://doi.org/10.1016/j.molmed.2014.11.010.
5. Chien S.-C., Chen C.-P., Liou J.-D. Prenatal diagnosis and genetic counseling of uniparental disomy // Taiwanese Journal of Obstetrics and Gynecology. 2022. Vol. 61, no. 2. P. 210–215. https://doi.org/10.1016/j.tjog.2022.02.006.
6. Nakka P., Smith S. P., O’Donnell-Luria A. H. et al. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population // The American Journal of Human Genetics. 2019. Vol. 105, no. 5. P. 921–932. https://doi.org/10.1016/j.ajhg.2019.09.016.
7. Сhen Q., Chen Y., Shi L. et al. Uniparental disomy: expanding the clinical and molecular phenotypes of whole chromosomes // Frontiers in Genetics. 2023. Vol. 14. https://doi.org/10.3389/fgene.2023.1232059.
8. Liehr T. Uniparental disomy (UPD) in clinical genetics: A guide for clinicians and patients. Springer, 2014. 192 p.
9. Вашукова Е. С., Тарасенко О. А., Козюлина П. Ю. и др. Оценка эффективности полногеномного неинвазивного пренатального тестирования для выявления редких хромосомных аномалий на основе опыта ФГБНУ «НИИ АГиР им. Д. О. Отта» // Медицинская генетика. 2022. Т. 21, № 11. С. 19–22.
10. Gonzales P. R., Andersen E. F., Brown T. R. et al. Interpretation and reporting of large regions of homozygosity and suspected consanguinity/uniparental disomy, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG) // Genetics of Medicine. 2022. Vol. 24, no. 2. P. 255–261. https://doi.org/10.1016/j.gim.2021.10.004.
11. Scuffins J., Keller-Ramey J., Dyer L. et al. Uniparental disomy in a population of 32,067 clinical exome trios // Genetics of Medicine. 2021. Vol. 23, no. 6. P. 1101–1107. https://doi.org/10.1038/s41436-020-01092-8.
12. Саженова Е. А., Лебедев И. Н. Стандарты молекулярно-генетической ДНК-диагностики болезней геномного импринтинга на примере синдромов Прадера-Вилли и Энгельмана // Медицинская генетика. 2015. Т. 14, № 9. С. 3–10.
13. Yamada M., Okuno H., Okamoto N. et al. Diagnosis of Prader-Willi syndrome and Angelman syndrome by targeted nanopore longread sequencing // European Journal of Medical Genetics. 2023. Vol. 66, no. 2. https://doi.org/10.1016/j.ejmg.2022.104690.
14. Holthöfer L., Diederich S., Haug V. et al. A case of an Angelmansyndrome caused by an intragenic duplication of UBE3A uncovered by adaptive nanopore sequencing // Clinical Epigenetics. 2024. Vol.16. https://doi.org/10.1186/s13148-024-01711-0.
15. Abdurashitov M. A., Tomilov V. N., Gonchar D. A. et al. Comparative analysis of RCGY sites methylation in three human cell lines // Epigenetic DNA diagnostics. 2019. https://doi.org/10.26213/ SE.2019.76.40116.
16. Mora-Márquez F., García-Olivares V., Emerson B. C. et al. ddRADseqTools: a software package for in silico simulation and testing of double-digest RADseq experiments // Molecular Ecology Resources. 2017. Vol. 17, no. 2. P. 230–246. https://doi.org/10.1111/1755-0998.12550.
17. Putri G. H., Anders S., Pyl P. T. et al. Analysing high-throughput sequencing data in Python with HTSeq 2.0 // Bioinformatics. 2022. Vol. 38, no. 10. P. 2943–2945. https://doi.org/10.1093/bioinformatics/btac166.
18. A resource for integrated genetical genomic analysis of the human Imprint Control Regions (ICRs). URL: https://humanicr.org/ (дата обращения: 15.01.2025).
19. Radtke M., Moch J., Hentschel, J. et al. altAFplotter: a web app for reliable UPD detection in NGS diagnostics // BMC Bioinformatics. 2024. Vol. 25. https://doi.org/10.1186/s12859-024-05922-3.
20. Jima D. D., Skaar D. A., Planchart A. et al. Genomic map of candidate human imprint control regions: the imprintome // Epigenetics. 2022. Vol. 17, no. 13. P. 1920–1943. https://doi.org/10.1080/15592294.2022.2091815.
магистрант
биоинформатик
врач – лабораторный генетик, кандидат медицинских наук, ведущий научный сотрудник
доктор биологических наук, заведующий отделом экспериментальной медицинской вирусологии, молекулярной генетики и биобанкинга
младший научный сотрудник
Сучко П.А., Некрасова Д.А., Донников М.Ю., Глотов О.С., Данилов Л.Г. МЕТОД СЕКВЕНИРОВАНИЯ И АЛГОРИТМ АНАЛИЗА ПРОФИЛЕЙ МЕТИЛИРОВАНИЯ НА ОСНОВАНИИ ДАННЫХ NGS-СЕКВЕНИРОВАНИЯ ДЛЯ ВЫЯВЛЕНИЯ ОДНОРОДИТЕЛЬСКИХ ДИСОМИЙ. Вестник СурГУ. Медицина. 2025;18(1):73-80. https://doi.org/10.35266/2949-3447-2025-1-9
Suchko P.A., Nekrasova D.A., Donnikov M.Yu., Glotov O.S., Danilov L.G. SEQUENCING METHOD AND ALGORITHM FOR ANALYZING METHYLATION PROFILES BASED ON NGS DATA TO IDENTIFY UNIPARENTAL DISOMY. Vestnik SurGU. Meditsina. 2025;18(1):73-80. (In Russ.) https://doi.org/10.35266/2949-3447-2025-1-9

![]()
![]()
628412, Тюменская обл., Ханты-Мансийский автономный округ – Югра, г. Сургут, ул. Энергетиков, 22
БУ ВО ХМАО-Югры «Сургутский государственный университет»
тел.: 8 (3462) 76-30-50
Email: science.jоurnals@surgu.ru













