


Содержание
Перейти к:
 А. С. Бурлаченко,
Д. А. Некрасова,
М. Ю. Донников,
А. М. Ермаков,
Е. С. Жданова,
Л. В. Коваленко,
Л. Г. Данилов,
О. С. Глотов
А. С. Бурлаченко,
Д. А. Некрасова,
М. Ю. Донников,
А. М. Ермаков,
Е. С. Жданова,
Л. В. Коваленко,
Л. Г. Данилов,
О. С. Глотов https://doi.org/10.35266/2949-3447-2025-2-11
Перейти к:
Использование современных методов секвенирования позволяет определять таксономическое разнообразие различных бактериальных сообществ. В особенности это позволяет оценить разнообразие штаммов бактерий, которые являются некультивируемыми. На текущий момент существует 2 поколения технологий секвенирования, позволяющие определять разнообразие микробного сообщества – 2‑е поколение (представлено Illumina и BGI) и 3‑е поколение (Nanopore и PacBio). Однако, так как данные технологии используют разные подходы к секвенированию, встает вопрос о воспроизводимости получаемых результатов. Проведенные исследования показали, что результаты, полученные с использованием методов секвенирования Illumina и Nanopore, различаются по индексам разнообразия и таксономическому разнообразию. Полученные результаты выявляют необходимость в разработке изменения методов пробоподготовки и выделения дезоксирибонуклеиновых кислот для получения однозначного состава микробного сообщества.
Бурлаченко А.С., Некрасова Д.А., Донников М.Ю., Ермаков А.М., Жданова Е.С., Коваленко Л.В., Данилов Л.Г., Глотов О.С. СРАВНЕНИЕ МЕТОДОВ ТАКСОНОМИЧЕСКОГО ОПРЕДЕЛЕНИЯ БАКТЕРИЙ ПО ПОСЛЕДОВАТЕЛЬНОСТИ ГЕНА 16S С ИСПОЛЬЗОВАНИЕМ ТЕХНОЛОГИЙ СЕКВЕНИРОВАНИЯ ILLUMINA И NANOPORE НА ПРИМЕРЕ НЕФТЯНЫХ ОБРАЗЦОВ. Вестник СурГУ. Медицина. 2025;18(2):81-87. https://doi.org/10.35266/2949-3447-2025-2-11
Burlachenko A.S., Nekrasova A.D., Donnikov M.Yu., Ermakov A.M., Zhdanova E.S., Kovalenko L.V., Danilov L.G., Glotov O.S. COMPARISON OF BACTERIA TAXONOMIC PROFILING METHODS VIA 16S RRNA GENE SEQUENCING USING ILLUMINA AND NANOPORE TECHNOLOGIES ON OIL SAMPLES. Vestnik SurGU. Meditsina. 2025;18(2):81-87. (In Russ.) https://doi.org/10.35266/2949-3447-2025-2-11
Нанопоровое секвенирование, представленное платформой Oxford Nanopore Technologies (ONT), является мощным инструментом для изучения сложных микробных сообществ в различных средах, включая нефтяные загрязнения. Данный метод основан на прохождении молекул ДНК через нанопоры, встроенные в мембрану, при этом изменение электропроводности позволяет идентифицировать нуклеотидные последовательности [1].
Ранее основным ограничением метода была высокая частота ошибок при определении последовательностей, что затрудняло точное филогенетическое и функциональное профилирование микроорганизмов. Однако за последние годы произошли значительные усовершенствования в химии секвенирования и алгоритмах basecalling, что позволило существенно повысить точность метода [1].
Одним из ключевых достижений стало внедрение новой химии пор (R9.4 и R10), которая обеспечила более стабильный сигнал и повысила точность считывания последовательностей. Современные алгоритмы basecalling, такие как Guppy, использующие рекуррентные нейронные сети, позволили увеличить точность одиночных ридов с 60 (в ранних версиях) до 96,5 % [2]. Благодаря этим усовершенствованиям стало возможным применение ONT для анализа сложных микробных сообществ, в том числе из экстремальных сред, таких как нефтяные пласты и загрязненные углеводородами участки.
Одним из ключевых преимуществ нанопорового секвенирования является возможность получения длинных прочтений, что критично при исследовании микробных сообществ и их идентификации до уровня вида. В частности, использование ONT MinION (Oxford Nanopore Technologies, Oxford, Великобритания) позволило анализировать рибосомные опероны почти полной длины, что обеспечивает точное филогенетическое определение видов и штаммов микроорганизмов [3]. В отличие от платформ коротких прочтений, таких как Illumina, ONT позволяет исследователям секвенировать ампликоны без фрагментации.
Несмотря на существующие ограничения, такие как необходимость дополнительных корректировок данных и зависимость от качества выделенной ДНК, технология ONT уже доказала свою эффективность в анализе микробных сообществ сложных сред. По сравнению с методами Illumina MiSeq V4–V5, нанопоровое секвенирование обеспечивает более детальное исследование состава бактериальных сообществ, позволяя идентифицировать не только родовой, но и видовой уровень таксономической принадлежности организмов [4].
Цель исследования – сравнение эффективности секвенирования участков 16S рРНК с использованием разных платформ секвенирования на примере образцов нефти.
Выделение ДНК. Выделение ДНК проводилось с использованием набора «Meta Soil» Raissoil» с заменой оригинальных буферов производителя на буферы из литературных данных в соответствии с протоколом из статьи [5].
Амплификация образцов для нанопорового секвенирования. В ходе исследования применяли праймеры, специфичные к полному оперону 16S рРНК [6] и гипервариабельным участкам V3–V4 [7]. Подбор осуществляли на основе анализа научной литературы и данных референсной базы Silva.
Для увеличения концентрации итогового продукта, ампликонов 16S рРНК для бактерий и коротких фрагментов V3–V4 у бактерий проводилась постановка полимеразной цепной реакции (ПЦР) с использованием набора Encyclo Plus PCR kit (Евроген, Россия). Для получения длинных фрагментов 16S рРНК ПЦР проводилась в два этапа. Для коротких фрагментов V3–V4 – в один.
Алгоритм ПЦР для 16S состоял из двух этапов амплификации. В первом раунде нарабатывался целевой ампликон. Во время второго этапа ПЦР производилось баркодирование образцов.
Все образцы разводились до итоговой концентрации не более 10 нг/мкл ДНК для уменьшения действия возможных клеточных ингибиторов в образце. Измерение концентрации ДНК перед и после амплификации проводили с помощью Qubit Fluorometer and Qubit RNA BR Assay Kit (Thermo Fisher Scientific, USA).
После образцы смешивали с реакционной смесью ПЦР (5X буфер, смесь dNTP (10 мМ), 50X Encyclo полимераза, деионизированной водой, смесь праймеров (10 мМ)). Полученная смесь использовалась для проведения амплификации с заданной программой: предварительная денатурация цепи ДНК при 95 °C 3 мин, цикл – денатурация 95 °C 30 сек, отжиг праймеров 61 °C 30 сек, элонгация 72 °C 3 мин, итоговая элонгация – 72 °C 5 мин. Количество циклов варьировалось от 35 до 40. Полученный продукт реакции визуализировался с помощью электрофореза в 1,5 % агарозном геле.
После первого этапа амплификации образцы подвергались очистке от неспецифических фрагментов и реагентов и концентрированию на спин-колонках с помощью набора Cleanup Mini (Евроген, Россия). Элюирование проводили в 15 мкл буфера.
Во втором раунде амплификации для увеличения концентрации целевого продукта и баркодирования образцов использовались ампликоны из первого раунда и смесь из деионизированной воды, свободной от нуклеаз, 10X Encyclo буфера для ПЦР, смеси dNTP (10 мМ), ПЦР праймера (10 мМ), 50X Encyclo полимеразы и смесь баркодов (10 мМ). Полученная смесь использовалась для проведения амплификации с заданной программой: предварительная денатурация цепи ДНК при 95 °C 3 мин, цикл – денатурация 95 °C 30 сек, отжиг праймеров 62 °C 30 сек, элонгация 72 °C 2 мин, итоговая элонгация 72 °C 5 мин, 15 циклов. Продукт реакции визуализировался с помощью электрофореза в 1,5 % агарозном геле.
В случае коротких фрагментов V3–V4 полимеразная цепная реакция проводилась в один этап. Создавалась общая реакционная смесь, включавшая в себя праймеры на вариативные регионы и баркоды (5X буфер, смесь dNTP (10 мМ), 50X Encyclo полимераза, деионизированной водой, смесь праймеров для V3–V4 (10 мМ), ПЦР праймера (10 мМ), смесь баркодов (10 мМ)). Полученная смесь использовалась для проведения амплификации с заданной программой: предварительная денатурация цепи ДНК при 95 °C 3 мин, этап наработки фрагментов V3–V4 – денатурация 95 °C 20 сек, отжиг праймеров 55 °C 30 сек, элонгация 72 °C 30 сек, 5 циклов, после шел этап пришивания баркодов: денатурация 95 °C 20 сек, отжиг праймеров 62 °C 30 сек, элонгация 72 °C 30 сек, 30 циклов, итоговая элонгация проводилась при 72 °C 10 мин. Полученный продукт реакции визуализировался с помощью электрофореза в 1,5 % агарозном геле.
По завершению всех этапов амплификации образцы подвергались очистке от реакционной смеси на спин-колонках Cleanup Mini (Евроген, Россия).
Нанопоровое секвенирование. Секвенирование производилось на приборе MinION (Oxford Nanopore Technologies, Oxford, Великобритания), использовался протокол для подготовки библиотеки Ligation Sequencing Kit (SQK-LSK109), с пропуском этапа баркодирования.
Предварительная очистка и удаление побочных фрагментов производилась на магнитных частицах MagPure A4 XP (Guangzhou Magen Biotechnology Co., Ltd, Китай) в соответствии с протоколом.
Бейсколлинг проводили с помощью программы Guppy (Oxford Nanopore Technologies, Oxford, Великобритания) версии 6.4.6 параллельно с процессом секвенирования.
Пробоподготовка образцов для оптического секвенирования. Ключевые этапы протокола включали в себя смесь 5 мкл буфера, содержащего высокоточную ДНК-полимеразу и соответствующую смесь дезоксирибонуклеозидтрифосфатов, 0,5 мкл прямого праймера 16S, 0,5 мкл обратного праймера 16S, 10 нг ДНК-матрицы и дополняли объем до 4 мкл воды Milli-Q. Кроме того, для контроля были подготовлены два отрицательных контрольных образца.
Получение ампликонов осуществлялось методом полимеразной цепной реакции. Процедура проведения реакции была следующей: начальное денатурирование проводилось при температуре 95 °C в течение 3 минут, после чего следовали 35 циклов, каждый из которых включал в себя 15-секундное денатурирование при 95 °C, отжиг при 55 °C в течение 30 секунд и удлинение фрагмента при 72 °C на протяжении 30 секунд. Завершалась реакция этапом при 72 °C в течение 5 минут, после чего температура была снижена до 4 °C и поддерживалась на этом уровне.
В текущем исследовании ДНК-библиотеки подвергали очистке от компонентов реакционной смеси ПЦР с использованием коммерческого набора для очистки ДНК AMPure XP, основанного на парамагнитных частицах, что позволяло эффективно удалять дезоксирибонуклеозидтрифосфаты (dNTPs), соли, праймеры и димеры праймеров. После завершения реакции ПЦР в каждую микропробирку с ДНК-библиотекой добавляли 16 микролитров парамагнитных частиц и 10 микролитров деионизированной воды типа Milli-Q для оптимизации удаления мелких фрагментов ДНК, инкубируя при комнатной температуре в течение пяти минут для обеспечения связывания ампликонов ДНК с частицами. Затем пробирки помещали на магнитный штатив и удаляли супернатант, к каждому образцу добавляли 200 микролитров свежеприготовленного 80 %-го этанола. После удаления этанола пробирки инкубировали при комнатной температуре в течение 3–5 минут для высушивания остаточных капель спирта. В окончательной стадии ДНК элюировали, добавляя 25 микролитров сверхчистой воды Milli-Q, а затем отбирали 23 микролитра элюата без магнитных частиц в чистые пробирки. Концентрация ДНК в полученных образцах была определена с использованием набора для количественного измерения двухцепочечную ДНК (dsDNA).
Индексы были лигированы к полученным ампликонам с использованием реакции ПЦР, в ходе которой применялись индексы Nextera XT Index 1 (N7XX) и Nextera XT Index 2 (S5XX). В реакционную смесь вводили 10 нг ДНК, 1 мкл каждого из индексов, что обеспечивало уникальную комбинацию двух различных типов последовательностей, а также 5 мкл буфера, содержащего высокоточную ДНК-полимеразу и смесь дезоксирибонуклеозидтрифосфатов KAPA HiFi HS RM. Остаточный объем доводили водой.
Программа амплификации была следующей: первый этап – нагревание до 94 °C в течение 3 минут, за которым следовало 8 циклов, включающих денатурацию при 94 °C в течение 30 секунд, отжиг при 57 °C в течение 30 секунд и завершение программы при 72 °C в течение 8 минут, после чего образцы оставляли при 4 °C.
Очистка амплифицированной ДНК снова проводилась с помощью набора AMPure XP, следуя ранее описанному протоколу. После очистки была выполнена оценка концентрации ДНК с использованием набора для количественного определения двухцепочечной ДНК (dsDNA). Далее образцы были пулированы с количеством ДНК каждого из образцов 30 нг.
Биоинформатическая обработка. Обработка результатов секвенирования проводилась с использованием среды программирования R (v. 4.4.3) [8]. Для фильтрации прочтений, удаления химер и определения таксономического разнообразия использовался пакет dada2 (v. 1.30.0) [9]. Для таксономической классификации ампликонных последовательностей использовали базу данных SILVA (v.138.2) [10], содержащую выровненные последовательности 16S рРНК с сопоставленной таксономией. Для последующих манипуляций использовались следующие пакеты: phyloseq (v. 1.46.0) [11] – для интеграции данных о таксономии и последовательностях с метаданными; vegan (v. 2.6–10) [12] – для расчета индексов α- и β-разнообразия. Статистическое сравнение индексов α-разнообразия проводилось с использованием теста Вилкоксона.
При обработке результатов секвенирования нами было обнаружено 111 родов при использовании секвенатора Oxford Nanopore и 89 родов при использовании технологии Illumina (рис. 1).
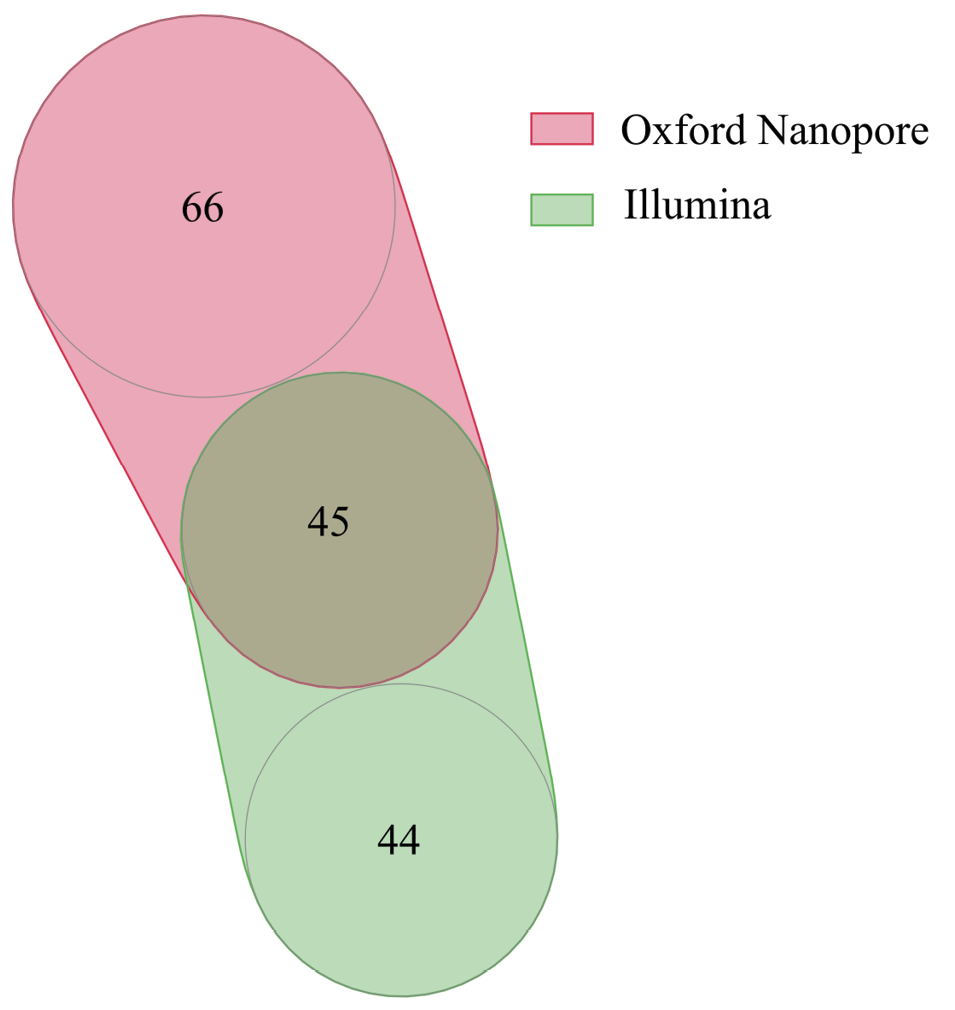
Рис. 1. Количество общих родов для разных платформ секвенирования
Примечание: составлено авторами.
Нами был рассчитан индекс биоразнообразия Шеннона для оценки представленности сообществ (рис. 2). Было показано наличие статистически значимых отличий (проведено сравнение с использованием теста Вилкоксона, p-value = 4.68e-08). Также стоит отметить, что секвенирование с использованием нанопоры приводило к обнаружению большего количества таксонов, что позволяет предположить возможность более точной детекции последовательностей.
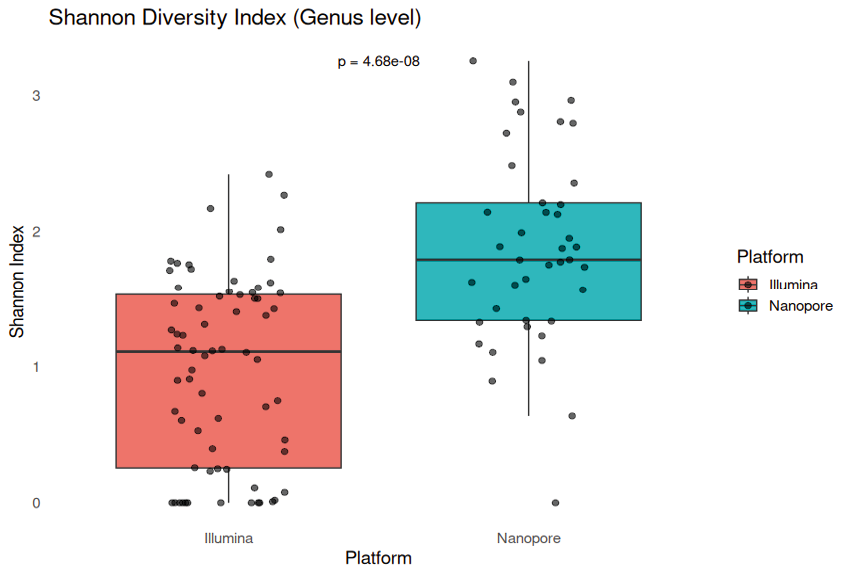
Рис. 2. Сравнение индекса Шеннона
Примечание: составлено авторами.
Проведенный нами PCoA-анализ, рассчитанный с учетом расстоянии Брея – Кертиса, показал частичное разделение микробиомных профилей, полученных с использованием исследуемых платформ (рис. 3). Анализ методом PERMANOVA подтвердил наличие статистически значимого отличия между группами (p-value = 0,001), что отражает различия в чувствительности к определению таксонов. Притом следует отметить, что повышенный уровень разнообразия наблюдался у образцов, отсеквенированных с использованием технологии Nanopore.
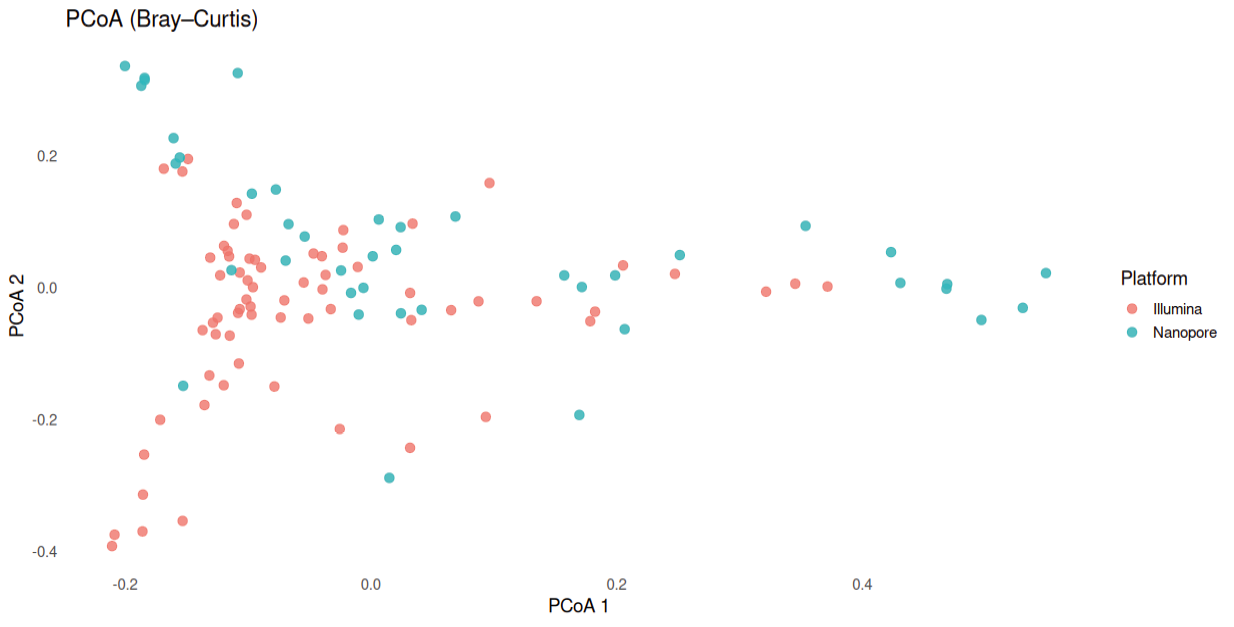
Рис. 3. Анализ бета-разнообразия образцов
Примечание: составлено авторами.
По результатам проведенного анализа можно сделать вывод о том, что особенности микробиомного состава нефтяных сообществ оказывают влияние на его оценку. Использование различных платформ секвенирования приводит к получению достаточно разнонаправленных результатов и для более полной картины необходимо сочетать различные методы секвенирования.
1. Rang F. J., Kloosterman W. P., de Ridder J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy // Genome Biology. 2018. Vol. 19.
2. Wick R. R., Judd L. M., Gorrie C. L. et al. Completing bacterial genome assemblies with multiplex MinION sequencing // Microbial Genomics. 2017. Vol. 3, no. 10. https://doi.org/10.1099/mgen.0.000132.
3. Karst S. M., Dueholm M. S., McIlroy S. J. et al. Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias // Nature Biotechnology. 2018. Vol. 36. P. 190–195. https://doi.org/10.1038/nbt.4045.
4. Kerkhof L. J., Dillon K. P., Häggblom M. M. et al. Profiling bacterial communities by MinION sequencing of ribosomal operons // Microbiome. 2017. Vol. 5.
5. Бурлаченко А. С., Данилов Л. Г., Глотов О. С. Оптимизация метода выделения бактериальной ДНК из нефтяных образцов // Бюллетень Оренбургского научного центра УрО РАН. 2024. № 4. С. 9.
6. Klindworth A., Pruesse E., Schweer T. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies // Nucleic Acids Research. 2012. Vol. 41, no. 1.
7. Neubeck A., Sun L., Müller B. et al. Microbial community structure in a serpentine-hosted abiotic gas seepage at the Chimaera Ophiolite, Turkey // Applied and Environmental Microbiology. 2017. Vol. 83, no. 12.
8. The R Project for Statistical Computing. URL: https://www.r-project.org/ (дата обращения: 01.04.2025).
9. Callahan B. J., McMurdie P. J., Rosen M. J. et al. DADA2: Highresolution sample inference from Illumina amplicon data // Nature Methods. 2016. Vol. 13. P. 581–583. https://doi.org/10.1038/nmeth.3869.
10. Quast C., Pruesse E., Yilmaz P. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools // Nucleic Acids Research. 2013. Vol. 41. P. D590–D596. https://doi.org/10.1093/nar/gks1219.
11. McMurdie P. J., Holmes S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data // PLoS ONE. 2013. Vol. 8, no. 4. https://doi.org/10.1371/journal.pone.0061217.
12. Oksanen J., Simpson G. L., Blanchet F. G. et al. vegan: Community Ecology Package. URL: https://CRAN.R-project.org/package=vegan (дата обращения: 01.04.2025).
младший научный сотрудник
кандидат фармацевтических наук, биоинформатик
врач-лабораторный генетик, кандидат медицинских наук, ведущий научный сотрудник
заведующий лабораторией
младший научный сотрудник
доктор медицинских наук, профессор, заведующая кафедрой патофизиологии и общей патологии, директор Медицинского института
младший научный сотрудник
доктор биологических наук, заведующий отделом экспериментальной медицинской вирусологии, молекулярной генетики и биобанкинга
Бурлаченко А.С., Некрасова Д.А., Донников М.Ю., Ермаков А.М., Жданова Е.С., Коваленко Л.В., Данилов Л.Г., Глотов О.С. СРАВНЕНИЕ МЕТОДОВ ТАКСОНОМИЧЕСКОГО ОПРЕДЕЛЕНИЯ БАКТЕРИЙ ПО ПОСЛЕДОВАТЕЛЬНОСТИ ГЕНА 16S С ИСПОЛЬЗОВАНИЕМ ТЕХНОЛОГИЙ СЕКВЕНИРОВАНИЯ ILLUMINA И NANOPORE НА ПРИМЕРЕ НЕФТЯНЫХ ОБРАЗЦОВ. Вестник СурГУ. Медицина. 2025;18(2):81-87. https://doi.org/10.35266/2949-3447-2025-2-11
Burlachenko A.S., Nekrasova A.D., Donnikov M.Yu., Ermakov A.M., Zhdanova E.S., Kovalenko L.V., Danilov L.G., Glotov O.S. COMPARISON OF BACTERIA TAXONOMIC PROFILING METHODS VIA 16S RRNA GENE SEQUENCING USING ILLUMINA AND NANOPORE TECHNOLOGIES ON OIL SAMPLES. Vestnik SurGU. Meditsina. 2025;18(2):81-87. (In Russ.) https://doi.org/10.35266/2949-3447-2025-2-11

![]()
![]()
628412, Тюменская обл., Ханты-Мансийский автономный округ – Югра, г. Сургут, ул. Энергетиков, 22
БУ ВО ХМАО-Югры «Сургутский государственный университет»
тел.: 8 (3462) 76-30-50
Email: science.jоurnals@surgu.ru













